OPENATOMManual: Modern Simulation Methods Applied to Chemistry, Physics and Biology
There are five types of files from which the OPENATOM
script-language interface reads commands, the
simulation setup file, the system setup file, molecular parameter/topology
files, the potential energy/pseudopotential parameter files and the
parallel decomposition parameter file. Coordinate and electronic state input files do
not contain commands but simply free format data.
In the simulation setup files, commands that drive a given simulation
are stated. For example, run 300 time steps of Car-Parrinello ab initio
molecular dynamics. In the system setup file, the
system is described. For example, 300 water molecules, two peptides,
three counter ions, thirty Kohn-Sham states etc. Topology files
contain information about the molecular connectivity and potential
energy/pseudopotential files contain information about the
interactions. The parallel decomposition
input file contains all the information about the parallel decomposition.
The idea behind the file division is that one often modifies the
simulation commands, occasionally the system setup commands but seldom the
topology, potential energy,pseudopotential or parallel decomposition
information.
The five file types help organization and transferability. For
example, many simulation command files can drive the same system setup file,
molecular parameter/topology files, potential energy/pseudopotential
parameter and parallel decomposition files.
There are three levels of grouping for the OPENATOM commands,
meta-keywords, keywords and key-arguments.
The first level, the meta-keywords, sort keywords into naturally
connected groups. The second level, the keywords, are specific
commands to the computer program. The third level, key-arguments,
are the arguments to the keywords. The syntax looks like:
~meta_keyword1[ \keyword1{keyarg1}
\keyword2{keyarg2} ].
~meta_keyword2[ \keyword1{keyarg1}
\keyword2{keyarg2} ].
The commands are case insensitive and can be specified in any order.
The present release of the code concentrates on the fine grained
parallel Car-Parrinello molecular dynamics (CPAIMD) computations
and scaling results are given in IBM J. Res. Dev. 52 (2007).
However, all the
bio-widget input parameters are supported and biomolecules can be
built. Some of the biomolecular functionality is not yet implemented
in the source. The QM/MM extension is a work in progress.
In order to run the code type:
OPENATOMMachine.x paraInfo.in simInput.in
where machine is the machine-type, simInput.in is the simulation input
file and paraInfo.in is the parallel decomposition input file.
All file names are arbitrary.
Warning: The code will warn the user and stop the run if it isn't happy
with the input. The designers felt that not running a
simulation was better than taking potentially inappropriate input
and forging ahead.
Also, the code doesn't like to overwrite files because the designers
have overwritten one too many useful files themselves and thought,
A file that big?!
It might be very useful,
But now it is gone.
Finally, many of the error messages are sometimes written in colloquial American English,
a product of too many late nights writing code. A language option has
not yet been implemented. The advisor may have to explain what
``Dude'' means. However, the code does have a lot of error
checking because there is nothing worse than
Wind catches lily,
Scatt'ring petals to the wind:
Segmentation fault.
which leads one to believe
Errors have occurred.
I can't tell where or why.
Lazy programmers.
This manual was, of course, also a late night accomplishment.
At this moment (May 2015),
- OpenAtom only supports norm-conserving pseudopotentials.
- The converter works for CPMD pseudopotentials. You may want to download CPMD pseudopotential library (visit
http://cpmd.org/download.)
-- Minjung Kim
Source codes (provided):
- make_mt_pot.f
- piny_make_grid_mt.f
Input files:
- potential file (e.g., Zn_mt_pot)
- wavefunction file (e.g., Zn_mt_wfn)
- PI_MD.MAKE : This file contains input information needed to run piny_make_grid_mt.f
Instruction:
- Open CPMD pseudopotential file you wish to convert (e.g., Zn_MT_PBE_SEMI.psp)
- Copy potential and wavefunction data from CPMD pseudopotential file (right after
&POTENTIAL and &WAVEFUNCTION) to potential (Zn_mt_pot) and wavefunction file (Zn_mt_wfn)
- Edit make_mt_pot.f
- Enter the names of your potential and wavefunction files into the appropriate ``open" statement (line 3 and 4)
- Enter the names of your angular momentum channel files you wish to create into the ``open" statement.
For example, if you have 3 angular momentum channels (
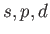 ), then Zn_l0, Zn_l1, Zn_l2
), then Zn_l0, Zn_l1, Zn_l2
- Compile make_mt_pot.f and run. It will create Zn_l0, Zn_l1, Zn_l2 files.
- Edit PI_MD.MAKE
The following informations are required:
- npts rmax lmax
- ZV
- alpha1 C1
- alpha2 C2
- l=0 file name
- l=1 file name
- l=2 file name
- ...
- l=lmax file name
npts: number of points on the ouput radial grid
rmax: the largest r value you on the radial grid
lmax: the highest angular momentum channel
ZV: the valence charge
alpha1, alpha2, C1, C2: parameters that determine the long-range part of the pseudopotential, assumed to be:
-C1*erf(alpha1*r)/r - C2*erf(alpha2*r)/r
We've been using alpha1=1.0 C1=1.0 and C2=0.0 so that the long-range part is -erf(r)/r
- Compile piny_make_grid_mt.f and run. It will create OpenAtom pseudopotential file.
Source code (provided):
Input file:
- input file (e.g., Cl_SG_BLYP_INPUT - see the example in the directory)
Instruction:
- Create an input file which includes the following information:
First line -comments
Zion
ZV (valence electrons)
LMAX
rloc
number of C, C1, C2, C3, C4
Number of s channels
rs
h0(1,1)
h0(2,2)
Number of p channels
rp
h1(1,1)
Number of d channels
Number of f channels
You can get these numbers from CPMD PP file. Or look up PRB 58, 3641 (1998) paper. All coefficients are tabulated.
- Run
make_goedecker.c. You are prompted for the name of the input file, and it will create PSEUDO.OUT.
Before running any type of ``CP'' (which means quantum in OPENATOM land) simulations supported by OPENATOM package, you first need to obtain minimized wavefunction files by performing cp_wave_min.
This document explains how to make input files for cp_wave_min with a system of one water (H O) molecule.
O) molecule.
-Minjung Kim
There are 7 different types of input files to run OPENATOM.
Let's create a file named water.input. The name of the input file is your choice. This file includes general information of the simulation such as, what type of simulation to perform, where to write the output files, where to find other input files, etc.
The simulation keyword file contains 9 subsections (i.e., meta-keywords):
~sim_list_def[ ]
~sim_cp_def[ ]
~sim_gen_def[ ]
~sim_class_PE_def[ ]
~sim_run_def[ ]
~sim_nhc_def[ ]
~sim_vol_def[ ]
~sim_write_def[ ]
~sim_pimd_def[ ]
It is not required to use all meta-keywords in the input file. For example, if you are not running your calculation with multiple beads, sim_pimd_def[] meta-keyword is unnecessary.
Each meta-keyword requires keywords and key-arguments. Complete information of keywords and key-arguments is found in OPENATOM website. This document explains a few keywords and its key-arguments that are necessary to run cp_wave_min.
Here is the example of water.input:
~sim_gen_def[
\simulation_typ{cp_wave_min}
\num_time_step{100000}
\restart_type{initial}
]
~sim_cp_def[
\cp_minimize_typ{min_cg}
\cp_restart_type{gen_wave}
]
~sim_run_def[
\cp_min_tol{0.001}
]
~sim_vol_def[
\periodicity{3}
]
~sim_write_def[
\write_screen_freq{3}
\write_dump_freq{100}
\in_restart_file{water.coords_initial}
\mol_set_file{water.set}
\sim_name{water}
\write_binary_cp_coef{off_gzip}
]
Short explanation for each keyword & argument:
- Simulation type is
cp_wave_min, which means minimizing wavefunction coefficients.
- It performs 100000 iterations. After 100000 iteration, it stops even if it does not converge.
- Restart type is
initial, which means it starts from scratch.
min_cg uses the conjugate gradient method for minimization.
gen_wave stands for generating wavefunction coefficients.
- Once the force of wavefunction reaches to the
cp_min_tol value, it stops the wavefunction minimization.
periodicity indicates the boundary condition. 3 means it is fully periodic.
write_screen_freq defines the frequency of writing information to an output file.
write_dump_freq determines how frequently write the wavefunction coefficients to STATES_OUT directory.
in_restart_file reads the name of coordinate input file. water.coords_initial will be read from ATOM_COORDS_IN directory.
mol_set_file reads the setup file named water.set.
sim_name sets the simulation name. In this example, water file will be generated at the end of the simulation.
off_gzip means the wavefunction coefficients are not binary and gzipped.
In the input file, keyword \mol_set_file has a key-argument water.set. This is a setup file. It gives the information of total number of electrons, types of of molecules (or atoms), pseudopotential database, etc.
The setup file contains 4 subsections (=meta-keywords):
~wavefunc_def[ ]
~molecule_def[ ]
~data_base_def[ ]
~bond_free_def[ ]
The first three meta-keywords are necessary. For ``CP'' simulations, we do not need the last meta-keyword. Below is what water.set may look like:
~wavefunc_def[
\nstate_up{4}\nstate_dn{4}\cp_nhc_opt{none}
]
~molecule_def[
\mol_name{Hydrogen}\mol_parm_file{./DATABASE/H.parm}
\num_mol{2}\mol_index{1}\mol_opt_nhc{mass_mol}
]
~molecule_def[
\mol_name{Oxygen}\mol_parm_file{./DATABASE/O.parm}
\num_mol{1}\mol_index{2}\mol_opt_nhc{mass_mol}
]
~data_base_def[
\inter_file{./water.inter}
\vps_file{./water.vps}
]
wavefunc_def defines the number of electrons. The number of states for spin-up and spin-down has to be the same.
molecule_def defines the name of the molecule and specify the parameter file (.parm files). In quantum simulations, molecule should be the individual atom.
data_base_def defines the name of interaction file (water.inter) and pseudopotential file (water.vps).
This file includes the information of atom coordinates and the size of the simulation cell. The name of the atom coordinate file (e.g., water.coords_initial) is defined in input file (see subsection above).
The coordinate file includes three parts:
[number of atoms] [number of beads] [number of path]
coordinates (in Å)
simulation cell information (in Å)
In this example, water.coords_initial may look like below:
3 1 1
0.757 0.586 0.0
-0.757 0.586 0.0
0.000 0.000 0.0
10 0 0
0 10 0
0 0 10
Make sure that the coordinates must follow the order defined in water.set file with \mol_index keyword, i.e., the first two coordinates for H, and the third coordinate for O.
The charm parameter file includes all options related to charm++. It can be an empty file. Usually, that is the best way to start. If OPENATOM complains about some key-arguments, change it accordingly. In our example, we will name this file as cpaimd_config.
In the setup file (water.set), we have specified two files: water.inter and water.vps. For ``CP'' calculations (remember in OPENATOM world, CP means quantum), water.inter is irrelevant. However, if this file does not exist, the code will complain, so it is necessary to create this file as well. water.vps indicates types of the pseudopotential, names of the pseudopotential file, the number of angular momentum, and local component of the pseudopotential. Here are examples of water.inter and water.vps.
water.inter file:
~inter_parm[\atom1{H}\atom2{H}\pot_type{null}\min_dist{0.1}\max_dist{12.9}]
~inter_parm[\atom1{O}\atom2{H}\pot_type{null}\min_dist{0.1}\max_dist{12.9}]
~inter_parm[\atom1{O}\atom2{O}\pot_type{null}\min_dist{0.1}\max_dist{12.9}]
water.vps file:
~PSEUDO_PARM[\ATOM1{H}\VPS_TYP{LOC}\N_ANG{0}\LOC_OPT{0}
\VPS_FILE{./DATABASE/H_BLYP_PP30.pseud}]
~PSEUDO_PARM[\ATOM1{O}\VPS_TYP{KB}\N_ANG{1}\LOC_OPT{1}
\VPS_FILE{./DATABASE/O_BLYP_PP30.pseud}]
In setup file, the topology keywords file is defined in \mol_parm_file keyword for each molecule. In this file, we specify which atoms consist of the molecule (in our case, atom is the molecule). The example of these files for our water system is below:
H.parm file:
~MOLECULE_NAME_DEF[\MOLECULE_NAME{Hydrogen}\NATOM{1}]
~ATOM_DEF[\ATOM_TYP{H}\ATOM_IND{1}\MASS{1.0}\CHARGE{1.0}
\cp_valence_up{0}\cp_valence_dn{0}\cp_atom{yes}]
O.parm file:
~MOLECULE_NAME_DEF[\MOLECULE_NAME{Oxygen}\NATOM{1}]
~ATOM_DEF[\ATOM_TYP{O}\ATOM_IND{1}\MASS{16.0}\CHARGE{6.0}
\cp_valence_up{4}\cp_valence_dn{4}\cp_atom{yes}]
The name of the pseudopotential files are indicated in potential parameter file (water.vps). Generating pseudopotential for OPENATOM can be found in the OPENATOM website.
The following commands maybe specified in the simulation setup file:
- ~sim_gen_def [16 keywords]
- ~sim_run_def [16 keywords]
- ~sim_nhc_def [28 keywords]
- ~sim_write_def [27 keywords]
- ~sim_list_def [14 keywords]
- ~sim_class_PE_def [25 keywords]
- ~sim_vol_def [4 keywords]
- ~sim_cp_def [29 keywords]
- ~sim_pimd_def [10 keywords]
A complete list of all commands employed including default
values are placed in the file:
~sim_write_def[ \sim_name_def{sim_input.out} ].
- \simulation_typ{cp,cp_pimd,...} :
This describes the type of simulation being performed.
- cp_wave_min : Minimize/optimize the Kohn-Sham wave functions (fixed nuclei)
If you don't have initial wave functions, you have to
use the gen_wave option below.
- cp_wave_min_pimd : Minimize/optimize the Kohn-Sham wave functions
over all the beads in preparation for a path
integral run (PIMD) with fixed nuclei. If you need
starting wave functions for all beads, you need gen_wave.
- cp : Car-Parrinello molecular dynamics (MD)
- cp_pimd : Car-Parinello path integral MD over all the beads
- bomd : Born-Oppenheimer molecular dynamics
- bomd_pimd : Born-Oppenheimer molecular dynamics for beads in path integral MD
-- planned for the near future--
cp_min : Minimize nuclei on the Born-Oppenheimer surface (provided by DFT)
- \ensemble_typ{nve,nvt,...} :
Contols the type of statistical ensemble used in the MD simulation
- nve : Run a (N,V,E) microcanonical MD simulation
- nvt : Run a (N,V,T) canonical simulation, temperature is controlled by
temperature keyword below
-- planned for the near future --
npt_i : (N,P,T) ensemble with volume scaling only (e.g., for liquids)
npt_f : (N,P,T) ensemble with full unit cell scaling (e.g., solids
with shear modes)
- \restart_type{initial,
restart_pos,restart_posvel,restart_all} :
Option controlling how the coordinate input file is read in:
- initial : Start using initial format
with atomic positions in Angstroms.
- restart_pos : Restart only the atomic positions.
Velocities and thermostat velocites (if used) are
sampled from a Maxwell distribution.
- restart_posvel : Restart both atmoic position
and atomic velocities. Thermostat velocities (if used)
are sampled from a Maxwell distribution.
- restart_all : Restart atomic positions,
velocities, and thermostat velocities (if used).
- \num_time_step{0} :
Total number of time steps (or iterations) of the simulation. Zero is
an error, it must be a positive integer.
- \time_step{1} :
Units of femtoseconds. This is the fundamental or smallest time
step. If no multiple time step integration is used, then this is the
time step. For example, in a cp simulation, this is the time step for
each CP step. In a minimization process, this controls the size of
the minimization step.
- \temperature{300} :
Atomic temperature. This is the temperature used for velocity rescaling,
velocity resampling, thermostatting, etc.
- \pressure{0} :
Pressure of system. Only used for simulations in the NPT ensembles.
-- planned for the near future --
\minimize_typ{min_std,min_cg,min_diis} :
Perform a minimization run using the method specified in curly brackets:
- min_std : Steepest descent.
- min_cg : Conjugate gradient.
- min_diis : Direct inversion in the
iterative subspace.
- \init_resmp_atm_vel{on,off} :
If turned on, initial atomic velocities are resampled from a
Maxwell distribution at temperature set by \temperature above.
Atomic masses are set elsewhere.
- \resmpl_frq_atm_vel{0} :
Atomic velocities are resampled with a frequency equal to the number of time steps
in the curly brackets. A zero indicates no resampling is to be done.
- \init_rescale_atm_vel{on,off} :
If turned on, atomic velocities are initially rescaled to preset
temperature. This does not randomize but just scales the velocities.
- \rescale_frq_atm_vel{0} :
Atomic velocities are rescaled to desired temperature
with a frequency equal to the number of time steps in the
curly brackets. A zero indicates no resampling is to be done.
- \init_rescale_atm_nhc{on,off} :
If turned on, atomic thermostat velocities are initially rescaled to
preset temperature. This is about recalling the Nose-Hoover Chain (NHC) velocities.
- \zero_com_vel{yes,no} :
If set to yes, the center of mass velocity is initialized to zero.
- \shake_tol{1.0e-6} :
Constraints not treated by the group constraint method are iterated to a
tolerance given in curly brackets.
- \rattle_tol{1.0e-6} :
Time derivative of constraints not treated by the group constraint
method are iterated to a tolerance given in curly brackets.
- \max_constrnt_iter{200} :
Maximum number of iterations to be performed on constraints before a
warning that the tolerance has not yet been reached is printed out and
iteration stops.
- \group_con_tol{1.0e-6} :
Group constraint method is iterated to a tolerance given in curly
brackets.
- \atm_nhc_tau_def{1000} :
Time scale (in femtoseconds) on which the thermostats should evolve.
Generally set to some characteristic time scale in the system.
- \init_resmp_atm_nhc{on,off} :
If turned on, atomic thermostat velocities will be initially resampled from
a Maxwell distribution.
- \atm_nhc_len{2} :
Number of elements in the thermostat chain. Generally 2-4 is adequate.
- \respa_steps_nhc{2} :
Number of individual Suzuki/Yoshida factorizations to be employed in
integration of the thermostat variables. Only
increase if energy conservation not satisfactory with default value.
- \yosh_steps_nhc{1,3,5,7} :
Order of the Suzuki/Yoshida integration scheme for the thermostat
variables. Only increase if energy conservation not satisfactory with
default value.
- \resmpl_atm_nhc{0} :
Atomic thermostat velocities will be resampled with a frequency specified
in the curly brackets. A zero indicates no resampling of thermostat
velocities.
- \respa_xi_opt{1,2,3,4} :
Depth of penetration of thermostat integration into multiple time step
levels. Larger numbers indicate deeper penetration. A one indicates
thermostat variables are updated at the beginning and end of
every step (XO option).
- \atm_isokin_opt{on:on/off}:
Thermostat the atoms using isokinetic NHC (on) or plain NHC (off).
- \cp_thermstats{on:on/off}:
Thermostat the electrons (on) or run them without control (off).
If ``on'', at least one isokinetic octopus NHC thermostat is placed on each
g-space plane of each state.
- \cp_num_nhc_iso{2}:
The number of tenticles in an isokinetic octopus NHC thermostat.
- \cp_nhc_chunk{1}:
The number of thermostats per g-space electronic state plane.
- \sim_name{sim_input.out} :
A complete list of all commands specified and defaults used is outputted to the specified file. This is useful for knowing exactly what is going on! (What you set and what the code set by default.)
- \mol_set_file{sim_atm_mol_set.in} :
Name of file containing specifying the system: atoms and molecules in the system, the number of bands for the electronic wave functions and k-points, and the databases for the pseudopotentials. Please see the HOWTO above for examples of what this contains.
- \write_binary_cp_coef{on,off,off_gzip,on_gzip} :
How the wave function coefficients are written to files.
- off : plain ASCII text format
- on : binary
- off_gzip : plain ASCII but compressed by gzip
- on_gzip : binary and compressed by gzip
- \read_binary_cp_coef{on,off,off_gzip,on_gzip} :
How the wave function coefficients are read from files, see above write for options.
- \write_dump_freq{1} :
The restart file of atomic coordinates and velocities, etc. will be
written with a frequency of iterations specified in curly brackets. Filename is specified by
next option out_restart_file.
- \out_restart_file{sim_restart.out} :
Name of the restart (dump) file containing final atomic
coordinates and velocities.
- \write_pos_freq{100} :
Atomic positions will be appended to the trajectory position file with
a frequency in iterations specified in curly brackets.
- \write_vel_freq{100} :
Atomic velocities will be appended to the trajectory velocity file with
a frequency specified in curly brackets.
- \in_restart_file{sim_restart.in} :
Name of the file containing initial atomic coordinates and
velocities for this run.
- \atm_pos_file{sim_atm_pos.out} :
Trajectory position file name.
- \atm_vel_file{sim_atm_vel.out} :
Trajectory velocity file name.
- \conf_file_format{binary,formatted} :
Trajectory files are written either in binary or formatted form.
To be implemented in the near future
- \write_screen_freq{1} :
Output information to screen will be written with a frequency specified
in curly brackets.
- \write_inst_freq{1} :
Instantaneous averages of energy, temperature, pressure, etc. will be
written to the instantaneous file with a frequency of iteration sspecified in curly
brackets.
- \instant_file{sim_instant.out} :
File to which instantaneous averages are to be written.
- \atm_force_file{sim_atm_force.out} :
Trajectory force file name.
- \conf_partial_file{sim_atm_pos_part.out} :
Partial trajectory position file name.
- \conf_partial_limits{1,0} :
Two numbers specifying the first and last atoms to be written to
the partial trajectory position file.
- \write_force_freq{1000000} :
Atomic forces are written to the trajectory force file with a
frequency specified in the curly brackets.
- \screen_output_units{au,kcal_mol,kelvin} :
Output information is written to the screen in units specified
in curly brackets.
- \neighbor_list{no_list,ver_list,lnk_list} :
Type of neighbor list to use. Optimal scheme for most systems:
\ver_list with a \lnk_lst update type.
- \update_type{lnk_lst,no_list} :
Update the verlet list using either no list or a link list.
- \verlist_skin{1} :
Skin depth added to verlet list cutoff. Needs to be optimized between
extra neighbors added and average required update frequency.
- \lnk_cell_divs{7} :
Number of link cell divisions in each dimension. Needs to be optimized
if used either as list type or update type.
- \ewald_kmax{7} :
Controls maximum length of k-vectors used in evaluating Ewald summation for
electrostatic interactions. The specified numbers gives the range of integer lattice vector coefficients along any dimension (from negative this number to this number).
- \ewald_alpha{7} :
Size of real-space damping (screening) parameter in Ewald sum. More specifically, the specified number here divided by the Volume to the 1/3 power give the 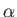 coefficient in the Ewald cutoff erfc(
coefficient in the Ewald cutoff erfc(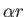 ) in real space.
) in real space.
- \inter_spline_pts{2000} :
Number of spline points used intermolecular interaction
potential and derivative. Namely this is about creating spline fits to erfc and similar functions.
- \periodicity{0,1,2,3} :
The type of periodic boundary conditions.
3 = full 3D periodicity
to be implemented in the near future:
0 = cluster
1 = wire
2 = slab
- \volume_tau{1000} :
Time scale of volume evolution
under constant pressure (NPT_I or NPT_F) in femtoseconds.
- \volume_nhc_tau{1000} :
Time scale of volume
heat bath evolution under constant pressure in femtoseconds.
- \cp_e_e_interact{on,off} :
Electron-electron interactions can be turned off (mainly for one electron problems or debugging).
Setting it off turns off exchange-correlation and Hartree energies and their contributions.
- \cp_dft_typ{lda,lsda,gga_lda,gga_lsda} :
Density functional exchange-correlation approximation type.
lda = local density approximation with no spin polarization.
lsda = lda but with spin.
gga_lda = generalized gradient approximation with no spin polarization.
gga_lsda = generalized gradient approximation with spin polarization.
- \cp_vxc_typ{pz_lda,pw_lda,pz_lsda} :
Base local density exchange correlation function type which can be corrected or overwritten by a gga choice (if selected). The local density types are Perdew-Zunger (pz), Perdew-Wang (pw), and Perdew-Zunger with spin (pz_lsda).
- \cp_ggax_typ{becke,pw91x,fila_1x,fila_2x,off} :
Gradient corrected exchange functional type. fila_1x and fila_2x are Filatov gradient corrections.
- \cp_ggac_typ{lyp,lypm1,pw91c,off} :
Gradient corrected correlation functional type. lyp is Lee-Yang-Parr, lypm1 is a modified lyp.
- \cp_grimme_vdw{on,off}:
Enables the Grimme van der Walls treatment of long-range interactions. Details are specified in the the DATA_BASE_DEF section of the input file specified by mol_set_file.
- \cp_norb{full_ortho,norm_only,off} :
Non-orthogonal orbitals option (see papers by Tuckerman-Hutter-Parr). Permits non-orthogonal wave functions.
- \cp_minimize_typ{min_std,min_cg} :
Minimization of electronic degrees of freedom. Steepest descent or conjugate gradient are the options but in practice please use min_cg (unless debugging).
- \cp_mass_tau_def{25} :
CP fictitious dynamics time scale in femtoseconds. This is equivalent to specifying the fictitious electronic mass for the fictitious CP dynamics of the electrons. If the kinetic energy of a g-vector 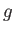 is
is
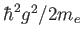 where
where 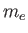 is the bare electron mass, then the oscillator frequency for that vector is
is the bare electron mass, then the oscillator frequency for that vector is
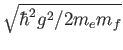 where
where 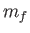 is the fictitious mass for that vector. The
is the fictitious mass for that vector. The 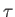 timescale is the inverse of this frequency which is set by this option. This means the masses for the different vectors (below a cutoff below) are set separately to ensure a single time scale.
timescale is the inverse of this frequency which is set by this option. This means the masses for the different vectors (below a cutoff below) are set separately to ensure a single time scale.
- \cp_energy_cut_def{2} :
Plane wave expansion cutoff
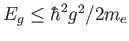 . Units are Rydbergs. One set of vectors is used for all k-points.
. Units are Rydbergs. One set of vectors is used for all k-points.
- \cp_mass_cut_def{2} :
Renormalize CP masses for
. Units are Rydbergs. This goes together with cp_mass_tau_def.
- \cp_fict_KE{1000} :
Fictitious initial kinetic energy and/or target kinetic energy (via thermostats) of the electrons during CP simulations specified by a temperature. Units are Kelvin.
- \cp_init_orthog{on,off} :
Orthonormalize the states at the start.
- \cp_orth_meth{gram_schmidt,lowdin} :
Orthonormalize using gram-schmidt or lowdin.
- \cp_restart_type{gen_wave,initial,
restart_pos,restart_posvel,restart_all} :
Start the run by generating initial wave functions.
- gen_wave = generates initial wave functions from atomic orbitals
- initial = read them from files
- restart_pos = same as initial
- restart_posvel = read coefficients and velocity coefficients from file (CP dynamics)
- restart_all = in addition to restart_posvel also reads thermostat velocities
- \cp_nonloc_ees_opt{on,off }:
Compute the non-local pseudopotential energy and forces using Euler
exponential spline interpolation (EES). This is 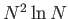 as opposed to
as opposed to 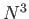 when off.
You should generally used EES unless perhaps you have an extremely small number of atoms
in your simulation cell when the standard algorithm might be faster. method is
only supposed for s-wave non-local pseudopotentials.
when off.
You should generally used EES unless perhaps you have an extremely small number of atoms
in your simulation cell when the standard algorithm might be faster. method is
only supposed for s-wave non-local pseudopotentials.
- \cp_eext_ees_opt{on,off }:
Compute the local pseudopotential energy and forces as well as the
reciprocal space part of the Ewald sum using EES. This makes things 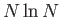 instead of
instead of 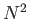 .
.
- \cp_pseudo_ees_order{6}:
Use a EES interpolation of specified order (polynomial interpolation order for Cardinal B-splines).
- \cp_pseudo_ees_scale{1.4}:
Use a g-space expanded by specified factor to perform the EES interpolation. This is a linear scaling of each dimension.
-- planned for the near future --
- \cp_ptens{on,off} :
Evaluate the pressure tensor.
To setup the initial configuration of a bunch beads, you should use the
src_external_conversion/spread_coords routines from an existing set of atomic positions.
- \path_int_beads{1} :
Number of path integral beads.
- \path_int_md_typ{staging,centroid} :
Path integral molecular dynamics type, staging or centroid/normal mode.
Staging gives the fastest equilibration.
- \path_int_gamma_adb{1} :
Path integral molecular dynamics type adiabaticity parameter.
Employed with the centroid option to give approxation to quantum dynamics.
- \num{1} :
Number of temperers
- \ens_opt{nvt} :
Ensemble in which you are tempering
- \statept_infile{statepoint.list} :
List of statepoints to be tempered.
- \screen_name{screen.out} :
Output name of any screen output for each temperer will be screen.out.#
- \switch_steps{1000} :
Number of steps between switches
- \output_directory{TEMPER_OUT} :
Output directory where files are stored.
The following commands maybe specified in the system setup file:
- ~molecule_def []
- ~wavefunc_def []
- ~bond_free_def []
- ~data_base_def []
- \mol_name{} :
The name of the molecule type.
- \num_mol{ } :
The number of this molecule type you want in your system.
- \num_residue{ } :
The number of residues in in this molecule type.
- \mol_index{2} :
This is the ``2nd'' molecule type defined in the system.
- \mol_parm_file{ } :
The topology of the molecule type is described in this file.
- \mol_text_nhc{ } :
The temperature of this molecule type. It may be different than the
external temperature for fancy simulation studies in the adiabatic limit.
- \mol_freeze_opt{none,all,backbone} :
Freeze this molecule to equilibrate the system.
- \hydrog_mass_opt{A,B} :
Increase the mass of type A hydrogens where A is
off,all,backbone or sidechain to B amu. This allows
selective deuteration for example.
- \hydrog_con_opt{off,all,polar} :
Constrain all bonds to this type of hydrogen in the molecule.
- \mol_nhc_opt{none,global,glob_mol,ind_mol,
res_mol,atm_mol,mass_mol} :
Nose-Hoover Chain option: Couple the atoms in this molecule to:
- none: no thermostats.
- global: the global thermostat.
- glob_mol: a thermostat for this molecule type.
- ind_mol: a thermostat for each molecule of this type.
- res_mol: a thermostat for each residue in each molecule.
- atm_mol: a thermostat for each atom in each molecule.
- mass_mol: a thermostat for each degree of freedom.
- \mol_tau_nhc{ } :
Nose-Hoover Chain time scale in femtoseconds for this molecule type.
- \nstate_up{1} :
Number of spin up states in the spin up electron density.
- \nstate_dn{1} :
Number of spin down states in the spin down electron density.
- \cp_tau_nhc{25} :
Nose-Hoover Chain coupling time scale.
- \num_kpoint{1} :
Number of kpoints. If it is not set (default), code assumes gamma point.
- \kpoint_file{pi_md.kpts} :
File where list of kpoints is stored. First line is number of kpoints followed
by list of kpoints and weights each on a separate line (ka kb kc wght) for a
file of nkpoint + 1 lines.
- \atom1_moltyp_ind{ } :
Index of molecule type to which atom 1 belongs.
- \atom2_moltyp_ind{ } :
Index of molecule type to which atom 2 belongs.
- \atom1_mol_ind{ } :
Index of the molecule of the specified molecule type
to which atom 1 belongs.
- \atom2_mol_ind{ } :
Index of the molecule of the specified molecule type
to which atom 2 belongs.
- \atom1_residue_ind{ } :
Index of the residue in the molecule to which atom 1 belongs.
- \atom2_residue_ind{ } :
Index of the residue in the molecule to which atom 2 belongs.
- \atom1_atm_ind{ } :
Index of the atom 1 in the residue.
- \atom2_atm_ind{ } :
Index of the atom 2 in the residue.
- \eq{ } :
Equilibrium bond length in Angstrom.
- \fk{ } :
Umbrella sampling force constant in K/Angstrom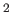 .
.
- \rmin_hist{ } :
Min Histogram distance in Angstrom.
- \rmax_hist{ } :
Max Histogram distance in Angstrom.
- \num_hist{ } :
Number of points in the histogram.
- \hist_file{ } :
File to which the histogram is printed.
- \inter_file{ } :
File containing intermolecular interaction parameters.
- \vps_file{pi_md.inter} :
File containing pseudopotential interaction parameters.
- \bond_file{pi_md.bond} :
File containing bond interaction parameters.
- \bend_file{pi_md.bend} :
File containing bend interaction parameters.
- \tors_file{pi_md.tors} :
File containing torsion interaction parameters.
- \onefour_file{pi_md.onfo} :
File containing onefour interaction parameters.
The following commands maybe specified in the molecular topology
and parameter files:
- ~ molecule_name_def []
- ~ residue_def []
- ~ residue_bond_def []
- ~ residue_name_def []
- ~ atom_def []
- ~ bond_def []
- ~ grp_bond_def []
- ~ bend_def []
- ~ bend_bnd_def []
- ~ tors_def []
- ~ onfo_def []
- ~ residue_morph []
- ~ atom_destroy []
- ~ atom_create []
- ~ atom_morph []
- \molecule_name{} :
Molecule name in characters.
- \nresidue{} :
Number of residues in the molecule.
- \natom{} :
Number of atoms in the molecule.
- \residue_name{} :
Residue name in characters.
- \residue_index{} :
Residue index number (this is the third residue in the molecule).
- \natom{} :
Number of atoms in the residue (after morphing).
- \residue_parm_file{} :
Residue parameter file.
- \residue_fix_file{} :
File containing morphing instructions for this residue if any.
- \res1_typ{} :
Residue name in characters.
- \res2_typ{} :
Residue name in characters.
- \res1_index{} :
Numerical index of first residue.
- \res2_index{} :
Numerical index of second residue.
- \res1_bond_site{} :
Connection point of residue bond in residue 1.
- \res2_bond_site{} :
Connection point of residue bond in residue 2.
- \res1_bondfile{} :
Morph file for residue 1 (lose a hydrogen, for example).
- \res2_bondfile{} :
Morph file for residue 2 (lose an halogen atom, for example).
- \res_name{} :
Name of the residue in characters.
- \natom{} :
Number of atoms in the residue
- \atom_typ{} :
Type of atom in characters.
- \atom_ind{} :
This is the nth atom in the molecule. The number is used to
define bonds, bends, tors, etc involving this atom.
- \mass{} :
The mass of the atom in amu.
- \charge{} :
The charge on the atom in ``e''.
- \valence{} :
The valence of the atom.
- \improper_def{0,1,2,3} :
How does this atom like its improper torsion constructed (if any).
- \bond_site_1{site,prim-branch,sec-branch} :
To what bond-site(s), character string, does this atom belong and where is
it in the topology tree structure relative to the root (the atom
that will bonded to some incoming atom).
- Atom to be bonded =root atom (0,0).
- 1st atom bonded to root atom (1,0).
- 2nd atom bonded to root atom (2,0).
- 1st atom bonded to 1st branch (1,1).
- 2nd atom bonded to 1st branch (1,2).
- \def_ghost1{index,coeff} :
If the atom is a ghost (these funky sites that TIP4P type
models have), it spatial position is constructed by taking linear
combinations of the other atoms in the molecule specified by the index
and the coef. A ghost can be composed of up to six other atoms
(ghost_1 ... ghost_6).
- \atom1{} :
Numerical index of atom 1.
- \atom2{} :
Numerical index of atom 2.
- \modifier{con,on,off} :
The bond is active=on, inactive=off, or constrained=con.
- Used to define a file which contains modifications to
a given residue.
- It has the same arguments as ~ residue_name_def[].
- Used to destroy an atom in a residue morph file.
- Uses the same key words as ~ atom_def[]
- Used to create an atom in a residue morph file.
- Uses the same key words as ~ atom_def[]
- Used to morph an atom in a residue morph file.
- Uses the same key words as ~ atom_def[]
The following commands maybe specified in the potential parameter files:
- ~ inter_parm []
- ~ bond_parm []
- ~ bend_parm []
- ~ tors_parm []
- ~ onfo_parm []
- ~ pseudo_parm []
- ~ bend_bnd_parm []
- \atom1{} :
Atom type 1 (character data).
- \atom2{} :
Atom type 2 (character data).
- \pot_type{lennard-jones,williams,aziz-chen,null} :
Potential type: Williams is an exponential c6-c8-c10. Aziz-chen has
a short range switching function on the vanderwaals part.
- \min_dist{} :
Minimum interaction distance.
- \max_dist{} :
Spherical cutoff interaction distance.
- \res_dist{} :
Respa Spherical cutoff interaction distance.
- \sig{} :
Lennard-Jones parameter.
- \eps{} :
Lennard-Jones parameter.
- \c6{} :
Williams/Aziz-Chen Vdw parameter.
- \c8{} :
Williams/Aziz-Chen Vdw parameter.
- \c9{} :
Williams/Aziz-Chen Vdw parameter.
- \c10{} :
Williams/Aziz-Chen Vdw parameter.
- \Awill{} :
Williams/Aziz-Chen parameter (
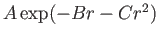 ).
).
- \Bwill{} :
Williams/Aziz-Chen parameter (
).
- \Cwill{} :
Williams/Aziz-Chen parameter (
).
- \rm_swit{} :
Williams/Aziz-Chen parameter controlling Vdw switching
(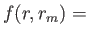 ).
).
- \atom1{} :
Atom type 1 (character data).
- \atom2{} :
Atom type 2 (character data).
- \pot_type{harmonic,power-series,morse,null} :
Potential type of bond.
- \fk{} :
Force constant of bond in K/Angstrom.
- \eq{} :
Equilibrium bond length in Angstrom.
- \eq_res{} :
Respa Equilibrium bond length in Angstrom.
- \alpha{} :
Morse parameter alpha in inverse Angstrom
(
![$\phi(r) = d_0[\exp(-\alpha(r-r_0))-1]^2$](img19.png) ).
).
- \d0{} :
Morse d0 in Kelvin
(
).
- \atom1{} :
Atom type in character data.
- \vps_typ{local,kb,vdb,null} :
Pseudopotential type: local, Kleinman-Bylander, Vanderbilt or null.
- \vps_file{} :
File containing the numerical generated pseudopotentials.
- \n_ang{} :
Number of angular momentum projection operators required.
- \loc_opt{} :
Which angular momentum pseudopotential is taken to be the local.
At present the code will build topologies for the user. However,
it will not build coordinates. Therefore, the user must provide
an initial input coordinate file( see \restart_type{initial}
and \in_restart_file{sim_restart.in})
The first line of the restart file must contain the number of atoms,
followed by a one, followed by the number of path integral beads
(usually 1). The code will grow beads and only P=1 need be provided
initially. The atoms positions then follow in the order specified by
the set file and the topology files. Finally, at the bottom of
the code the 3x3 simulation cell matrix must be specified.
The unit is Angstrom.
Example:
The molecular set file specifies 25 water molecules as molecule
type 1 and 3 bromine atom as molecule type 2. The water parameter file is
written as OHH, i.e. oxygen is atom 1, hydrogen1 is atom 2, hydrogen2 is
atom 3. Therefore, the input file should have 25 OHH coordinates followed
3 bromines. If the box is a square 25 angstrom on edge
then the last three lines of the file should be
25 0 0
0 25 0
0 0 25
The following commands maybe specified in the parallel decomposition file:
- ~ charm_conf_gen_def[ ]
- ~ charm_conf_rho_def[ ]
- ~ charm_conf_state_def[ ]
- ~ charm_conf_PC_def[ ]
- ~ charm_conf_NL_def[ ]
- ~ charm_conf_map_def[ ]
If no commands are set, the charm++ driver will decide values based
on system size and processor number and architecture. Below, the
name nstate refers to the number of KS states and the name nproc
refers to the number of processors to be used.
- \useCommlib{on:on,off } :
Turn on/off Charm communication library use. Default on
- \useCommlibMulticast{ } :
Turn on/off Charm multicast communication library use. Default on
- \atmOutput{on:on,off } :
Turn on/off atom output. Default on
- \gExpandFactRho{1 } :
Expands the g-space parallelization of the electron density beyond
the number of planes by the specified non-integer multiplicative factor.
- \rhoGHelpers{1 } :
Expands the g-space parallelization of the local pseudopotential
evaluation by the integer increment.
- \rhoRsubplanes{1 } :
Expands the r-space parallelization of the exchange correlation and
local pseudopotential energies by the integer increment.
The latter only matters if EES is on.
- \rhoLineOrder{skip:skip,none,random } :
Static Load balance scheme for density g-space parallelization.
- \nchareHartAtmT{1 :
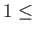
 natmTyp} :
natmTyp} :
Decomposition for the local pseudopotentil energy for use with the
EES method.
- \rhoSubPlaneBalance{off:on/off } :
When rhoRsubplanes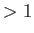 , balance the communication.
, balance the communication.
- \rhoGToRhoRMsgCombine{off:on/off } :
When rhoRsubplanes, combine messages.
- \dataPath{./STATES } :
The directory in which the electronic state input files are
stored. If the ``gen_wave'' option is selected, the dataPath
command is ignored.
- \dataPathOut{./STATES_OUT } :
The directory in which the electronic state output files are stored.
- \gExpandFact{1.0 } :
Expands the g-space parallelization of the electronic states beyond the number of planes by
the specified non-integer multiplicative factor. If the torus map option is
employed, the product must be a power of 2, for example
(gexpandfact=2)x(nplane_state=4)=8.
- \stateOutput{on: on/off } :
Turn the electronic state input on and off.
- \sGrainSize{ nstate} :
Integer smatrix decomposition parameter. The default value is number of
electron states, e.g. the matrix is not decomposed.
- \orthoGrainSize{ nstate} :
Integer lowdix decomposition parameter. The default value is number of
electron states, e.g. the matrix is not decomposed. This parameter
must be sGrainSize and
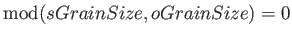 .
.
- \numChunks{ 1 } :
Increase the g-space decomposition by the integer factor.
- \phantomSym{ on:on/off} :
Add some extra parallelization for Smatrix computations.
- \useOrthoHelpers{ off:on/off} :
Add some extra parallelization for Lowdin orthogonalization.
Use only if (nstates/orthograinsize)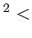 nproc.
nproc.
- \invsqr_tolerance{1e-15 } :
Tolerence on the iterative matrix square root method.
- \invsqr_max_iter{10 } :
Maximum number of iteration for the matrix square root method.
- \numSfGrps{ 1} :
Decompose the non-local computation into the specified number of groups.
- \numSfDups{ } :
Duplicate the evaluation of the structure factor for the
non-local computation the specified number of times.
- \launchNLeesFromRho{rs:rs,rhor,rhog } :
Specify the launch point for the Non-local EES computation.
- \Gstates_per_pe{nstates } :
If torusMap is off, specify the g-space state decomposition.
- \Rstates_per_pe{nstates } :
If torusMap is off, specify the r-space state decomposition.
- \torusMap{on: on/off} :
If your machine is a torus, this mapping scheme gives optimal performance.
- \loadMapFiles{off:on/off } :
Load pregenerated map files.
- \dumpMapFiles{off:on/off } :
Create map files.
- \fakeTorus{off:on/off } :
If your machine is a NOT torus, you can still use torus mapping.
- \torusDimX{ nproc} :
Fake Torus dimension along x in processors/node
- \torusDimY{1 } :
Fake Torus dimension along y in processors/node
- \torusDimZ{1 } :
Fake Torus dimension along z in processors/node
- \torusDimNX{1 } :
Fake Torus dimension along x in nodes
- \torusDimNY{1 } :
Fake Torus dimension along y in nodes
- \torusDimNZ{ 1} :
Fake Torus dimension along z in nodes
OPENATOMManual: Modern Simulation Methods Applied to Chemistry, Physics and Biology
This document was generated using the
LaTeX2HTML translator Version 2008 (1.71)
Copyright © 1993, 1994, 1995, 1996,
Nikos Drakos,
Computer Based Learning Unit, University of Leeds.
Copyright © 1997, 1998, 1999,
Ross Moore,
Mathematics Department, Macquarie University, Sydney.
The command line arguments were:
latex2html -split 2 -local_icons OpenAtomFullDoc.tex
The translation was initiated by Sohrab Ismail-Beigi on 2015-09-17
Sohrab Ismail-Beigi
2015-09-17